
by City of Hope National Medical Center
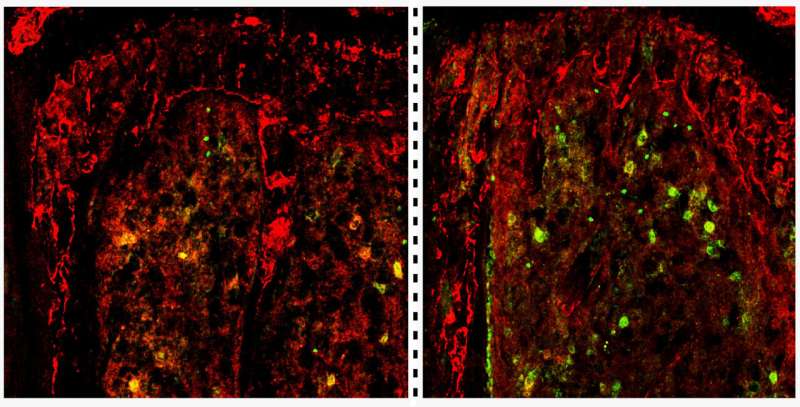
Scientists at City of Hope, one of the largest cancer research and treatment organizations in the United States, have identified how low levels of the TET2 gene fuel the rapid growth of acute myeloid leukemia in animal models. Cell Stem Cell recently published the study.
A team led by Jianjun Chen, Ph.D., the Simms/Mann Family Foundation Chair in Systems Biology at Beckman Research Institute of City of Hope, found that TET2 deficiency sets off a cascade of biochemical changes that enhance the bone marrow cancer's ability to spread. These changes include:
Driving the movement of malignant stem cells from the bloodstream to the bone marrow region where they originated. This home microenvironment, called a niche, protects the cells' survival and ability to divide and self-replicate.
Increasing the expression of a protein called TSPAN13 that signals leukemia stem cells to travel back to the bone marrow niche
Leading to the buildup of a methylated form of the RNA base cytosine that enhances TSPAN13 messenger RNA's stability, resulting in the increased expression of TSPAN13 protein
Activating a signaling pathway called the TSPAN13/CXCR4 axis that increases malignant stem cells' return (i.e., homing) to the bone marrow niche and self-replication, thereby leading to the rapid development of leukemia.
By expanding understanding of the multiple ways that TET2 influences the development of acute myeloid leukemia, the discovery points to new potential therapeutic targets for treating the disease.
"This study provides novel insights into the cellular and molecular mechanisms underlying the development of acute myeloid leukemia," Chen said. "Our findings highlight the therapeutic potential of reactivating TET2 signaling in patients with TET2 mutations or transcriptional suppression. Equally exciting, this strategy could be applied to other types of cancer that feature TET2 deficiency."

Acute myeloid leukemia is distinguished by the rapid division and spread of immature leukemia stem cells. More than half of patients relapse, and the five-year survival rate is only 30%. Figuring out how to destroy these cells is crucial to effectively treating the disease.
TET2 deficiency cooperates with leukemia-related oncoproteins resulting from chromosomal abnormalities or gene mutations to drive the development of leukemia and enhance malignant stem cells' ability to divide and spread. Until now, however, the cellular and molecular mechanisms underlying these processes have remained murky.
In analyzing data from the Cancer Genome Atlas, Chen and his colleagues found that lower expression or mutation of TET2 was associated with a poor prognosis and shorter overall survival rates for patients. Compared to healthy control subjects, TET2 expression was significantly suppressed in acute myeloid leukemia patients. This led the team to investigate the clinical relevance of TET2 in the development of the disease.
More information: Yangchan Li et al, TET2-mediated mRNA demethylation regulates leukemia stem cell homing and self-renewal, Cell Stem Cell (2023). DOI: 10.1016/j.stem.2023.07.001
Journal information: Cell Stem Cell
Provided by City of Hope National Medical Center
by Katie Neith, City of Hope National Medical Center
In a recent study led by Lei Jiang, Ph.D., an assistant professor of molecular and cellular endocrinology, a team of researchers from City of Hope and the University of Texas Southwestern Medical Center, found a potential new target for treating patients with metastatic cancer. Their findings were published in the August 29 issue of the journal Cell Reports.
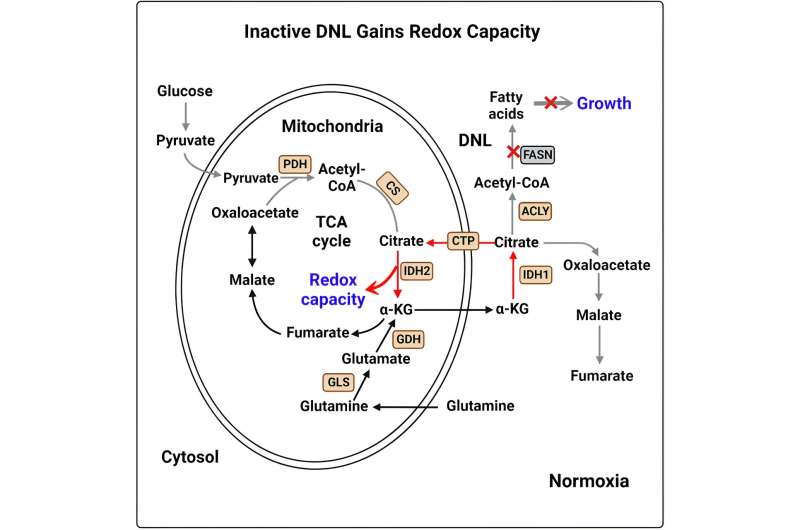
The goal of the team's study was to elucidate the role of reductive carboxylation in redox metabolism, a process believed to be important for metastatic cancer. Reductive carboxylation is best known as a metabolic pathway that provides a molecule called acetyl-CoA so that it can be turned into lipids, which is mediated by fatty acid synthase (FASN). The FASN-mediated lipogenesis process supports rapid growth in most proliferating cancer cells, and increased FASN expression has been viewed as a metabolic feature of cancer cells. Thus, FASN is considered a potential target to block tumor growth.
In their new paper, Jiang and colleagues used lung cancer cells to show that FASN inhibition induces reductive carboxylation, which further increases redox capacity (a process central to metabolism) in metastatic cancer cells. In this setting, reductive carboxylation induces a net cytosol-to-mitochondria citrate flux in FASN-deficient cells. This was surprising because citrate flux across mitochondria—one way that cancer cells gain energy—has been previously known to only go in the mitochondria-to-cytosol direction. Importantly, blocking this cytosol-to-mitochondria citrate flux effectively induces oxidative stress and cell death in metastatic cancer cells.
Previous work by Jiang and others found that adaptation to anchorage independence in cancer cells (meaning they can proliferate without another surface to anchor with) requires a fundamental metabolic change in the citrate metabolism—a lipogenic precursor for de novo lipogenesis—via reductive carboxylation to suppress oxidative stress. However, the role of fatty acid synthase (FASN), a critical lipogenic enzyme, in reductive carboxylation during metastatic progression was unclear prior to the new study.
Building on prior research, this study provides additional data to support Jiang and his team's belief that targeting the cytosol-to-mitochondria citrate flux process can be an effective therapy for treating cancer patients with metastasis. Since the role of FASN in metastasis is context dependent, the team plans to test whether a similar mechanism exists in other cancer types beyond lung cancer.
More information: Wenting Dai et al, FASN deficiency induces a cytosol-to-mitochondria citrate flux to mitigate detachment-induced oxidative stress, Cell Reports (2023). DOI: 10.1016/j.celrep.2023.112971
Journal information: Cell Reports
Provided by City of Hope National Medical Center





Post comments